
Welcome to the Gene Scene! Each week, we will explore a gene from the ACMG Secondary Findings list—genes identified by the American College of Medical Genetics and Genomics as having clear, actionable health implications. These genes are included because they’re linked to serious but preventable or manageable conditions when identified early. Here, we focus on the condition that led to the gene’s inclusion on the list, providing clear, relevant information that supports your clinic. To subscribe to the Gene Scene, contact your local GSL or send a request to info@ambrygen.com.
To access the Gene Scene archives, visit our blog.
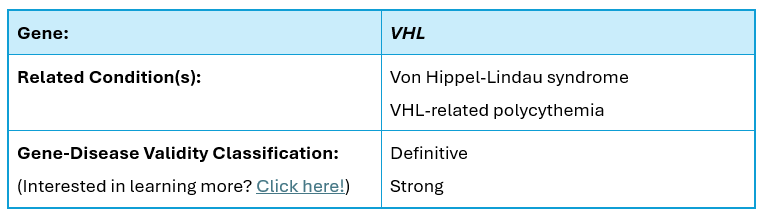
Clinical Phenotype Summary:
The VHL gene (NM_000551.3), which contains 3 coding exons and is located on chromosome 3p25.3, encodes the von Hippel-Lindau disease tumor suppressor protein. Pathogenic variants in this gene are known to cause Von Hippel-Lindau syndrome (VHLS), an autosomal dominant cancer predisposition syndrome with a 20% de novo rate. Additionally, pathogenic variants in this gene cause VHL-related polycythemia, which is inherited in an autosomal recessive fashion.
VHLS is characterized by:
• Renal tumors,
• Pheochromocytomas (PCCs),
• Retinal angiomas,
• Central nervous system (CNS) hemangioblastomas,
• Endolymphatic sac tumors,
• Pancreatic cysts, and
• Neuroendocrine tumors.
The condition can be divided into two primary types based on clinical features: VHL type 1 and VHL type 2. VHL type 2, which is further divided into 2A, 2B, and 2C subgroups, is differentiated from type 1 by a high risk of PCCs. VHL type 2A is characterized by PCC, retinal angiomas, and CNS hemangioblastomas, while type 2B is characterized by these features as well as pancreatic lesions and a high renal cell carcinoma (RCC) risk. VHL type 2C is characterized by risk of PCCs only.
Depending on disease subtype, VHLS-associated lifetime risks of RCC and PCCs are estimated at 25-70% and 10-26%, respectively. VHLS-associated PCCs are frequently bilateral, but rarely malignant.
Loss of function has been reported as the mechanism of disease for VHLS.
VHL-related polycythemia is characterized by:
• increased red blood cell mass,
• increased hematocrit, and
• increased hemoglobin with normal oxygen affinity, which may manifest with headache, dizziness, and dyspnea as well as an increased risk for peripheral arterial and venous thrombosis and cerebrovascular events.
VHL-related polycythemia may result from hypersensitivity to erythropoietin (EPO; primary erythrocytosis), increased levels of EPO (secondary erythrocytosis), or both. Age of onset is variable, typically occurring between early infancy and early adulthood.
Biallelic loss of function, with at least one hypomorphic allele, has been reported as the mechanism of disease for VHL-related polycythemia.
Unique Considerations:
Some variants in VHL are only associated with autosomal recessive VHL-related polycythemia and not autosomal dominant von Hippel-Lindau syndrome. Individuals found to have a single variant that is only associated with the autosomal recessive condition will have a report stating the individual tested is a “Carrier.” The variant details paragraph will include information on the associated condition.
Note: This information was reviewed at the time of publication for clinical accuracy, but advances in genetics happen quickly. For the most current information, please reach out to your Ambry representative or contact us at info@ambrygen.com.
Clinical Resources:
Clinician Management Resource for VHL and Understanding Your Positive VHL Genetic Test Result
Ambry Knows Genes:
Peer-Reviewed Publications:
• VHL mosaicism: the added value of multi-tissue analysis (March 2022)
Scientific Presentations:
Scientific Posters:
• VHL Information Sharing International Consortium (VISION): A ClinGen Expert Panel to Evaluate VHL Gene-Specific Criteria for Variant Interpretation (ACMG 2019)
• VHL Information Sharing International Consortium (VISION): A ClinGen Expert Panel to Evaluate VHL Gene-Specific Criteria for Variant Interpretation (ASHG 2018)
Citations:
• Lonser R et al. Lancet. 2003 Jun 14;361(9374):2059-67 PMID: 12814730
• Maher E et al. Eur J Hum Genet. 2011 Jun;19(6):617-23 PMID: 21386872
• Welander J et al. Endocr Relat Cancer. 2011 Dec 1;18(6):R253-76 PMID: 22041710
• Fishbein L and Nathanson K. Cancer Genet. 2012 Jan-Feb;205(1-2):1-11 PMID: 22429592
• Pastore Y et al. Am J Hum Genet. 2003 Aug;73(2):412-9 PMID: 12844285
• Gordeuk VR et al. Blood. 2004 May 15;103(10):3924-32 PMID: 14726398
• Hudler P and Urbancic M. Genes. 2022 Feb 17;13(2):362 PMID: 35205407
Ambry Genetics Gene-Disease Validity Scheme
Each week, we explore a gene from the ACMG Secondary Findings list—genes identified by the American College of Medical Genetics and Genomics as having clear, actionable health implications. These genes are included because they’re linked to serious but preventable or manageable conditions when identified early.
To learn more about the ACMG Secondary Findings list, click here.



