As a research fellow in the Cancer Genetics and Prevention group at Dana-Farber Cancer Institute—trained as a medical oncologist in Spain and specializing in hereditary cancer and translational genomics—I am often asked how to counsel patients who carry rare pathogenic variants, particularly when evidence is limited or outdated. Few scenarios illustrate this challenge more clearly than germline pathogenic or likely pathogenic variants (gPVs) in SMARCA4, a gene strongly linked to small cell carcinoma of the ovary, hypercalcemic type (SCCOHT), an exceptionally aggressive ovarian cancer.
Today, I’m excited to share results from our newly published study in Gynecologic Oncology, where we analyzed a decade of clinical testing data to better characterize cancer patterns among individuals with SMARCA4 gPVs, trying to address long-standing gaps in counseling, surveillance, and prevention.
Why This Matters: A Rare Tumor with an Urgent Need for Clarity
SCCOHT is extremely rare—representing less than 0.01% of all ovarian cancers1, 2—but it carries a devastating prognosis and typically affects teenagers and young adults3, 4. Prior studies have shown a strong association between SMARCA4 loss-of-function pathogenic variants and SCCOHT5-9, yet:
• penetrance estimates have been limited,
• most published data come from SCCOHT-enriched families,
• age-at-diagnosis information is inconsistent, and
• guidance on risk-reducing surgery is highly heterogeneous.
For patients and families carrying SMARCA4 gPVs, this uncertainty complicates key decisions: How high is my risk? How early can cancer develop? Should I consider risk-reducing salpingo-oophorectomy—and at what age?
Our study provides critical real-world insights to help answer these questions.
What We Did: A Comprehensive Look at SMARCA4 across 10 Years of Testing
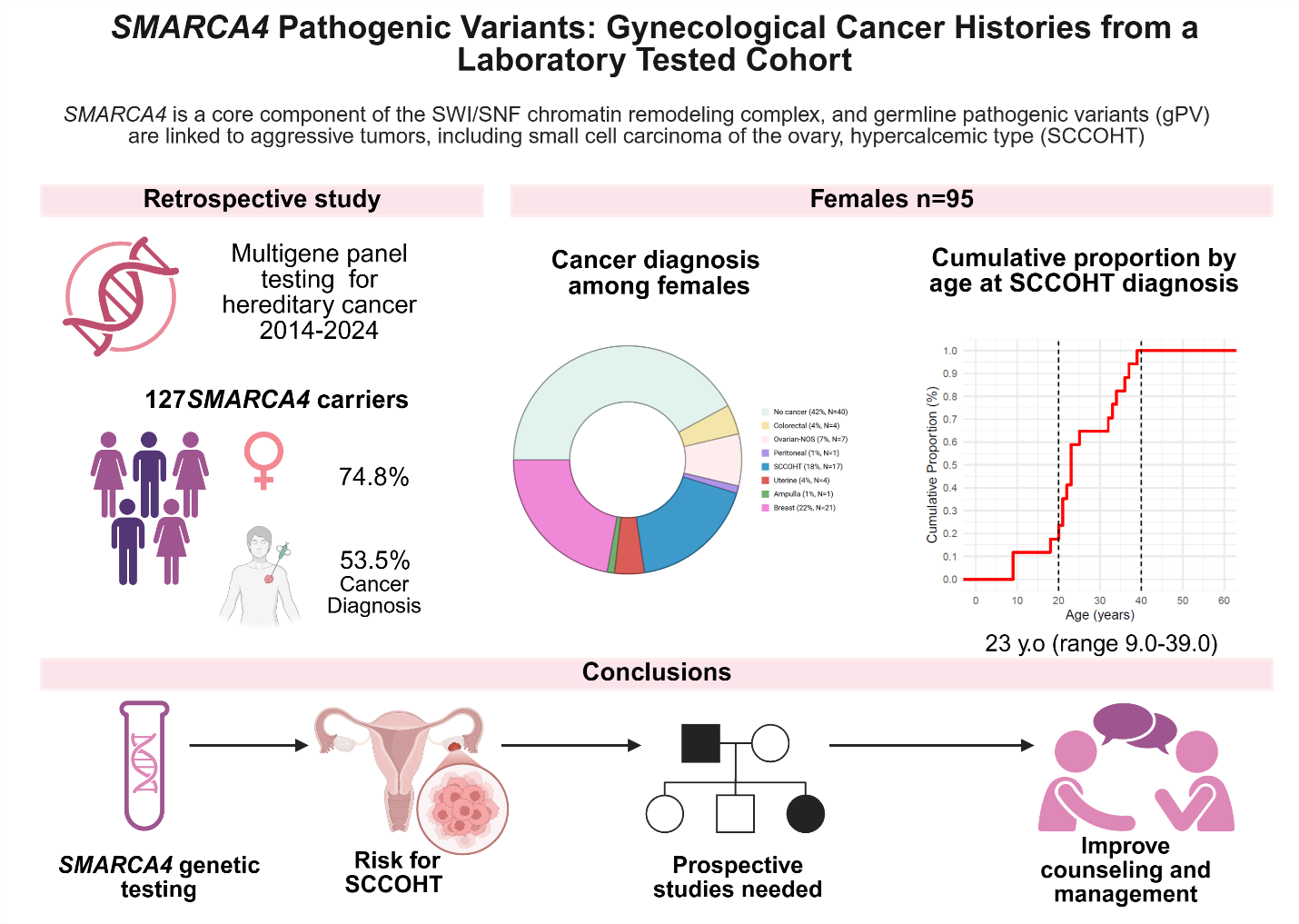
We conducted a retrospective review of 127 individuals with SMARCA4 germline loss-of-function variants identified through multigene panel testing between 2014 and 2024. This included probands and their relatives, enabling a broader and less biased view of cancer history compared to SCCOHT-only cohorts.
Key features of the cohort:
• 74.8% (n=95) were female
• 53.5% (n=68) had a personal history of cancer
• Median age at germline testing: 46 (31.0-59.0)
• Only loss-of-function variants (nonsense, frameshift, splice-site, or deletions) were included
Variants associated with Coffin-Siris syndrome or concurrent gPVs in other ovarian cancer genes (like BRCA1/2, ATM, RAD51D, MSH6) were excluded to isolate the effects of SMARCA4.
What We Found: SCCOHT Develops Early—and Only Early
Our results demonstrate a striking and consistent pattern:
• 17.9% (n=17) of females with a SMARCA4 gPV had SCCOHT.
• Median age at SCCOHT diagnosis was only 23 years (range 9–39).
• All SCCOHT cases occurred before age 40.
Among younger women (≤50 years), SCCOHT accounted for almost 30% of all cancer diagnoses.
In contrast, no cases were observed after age 40, consistent with patterns shown in the cumulative age curves (Fig. 3A and 3B, page 4)
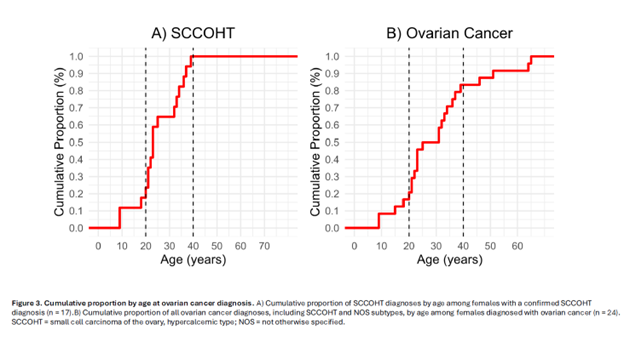
These data affirm that SCCOHT risk is heavily concentrated in adolescence and early adulthood, with implications for both surveillance and timing to consider and discuss preventive surgery.
Implications for Clinical Care
1. SMARCA4 should be routinely included in hereditary ovarian cancer panels for young individuals, especially those diagnosed before age 40.
Our data support existing recommendations highlighting SMARCA4 in the evaluation of early-onset ovarian cancer.
2. Risk-reducing salpingo-oophorectomy should be discussed early.
Given that all SCCOHT cases occurred before age 40, consideration of preventive surgery in early adulthood may be appropriate for selected individuals—balanced with fertility preservation, psychosocial impact, and emerging therapeutic options.
3. Current surveillance strategies are insufficient.
Pelvic ultrasound and MRI have shown limited efficacy for SCCOHT detection. Some guidelines propose childhood imaging for RTPS2, but its value remains unclear for ovarian cancer screening10.
4. Management must be individualized.
Importantly, 82% of women in our cohort did not develop SCCOHT, underscoring the need for nuanced counseling rather than a one-size-fits-all strategy.
Looking Forward
SCCOHT has poor outcomes with conventional therapy, but evolving approaches—including immunotherapy—are beginning to show promise4. Because of the rarity of this cancer, ongoing collaborative efforts such as the SCCOHT-SMARCA4 Registry and Biobank are essential to accelerate discovery and refine clinical recommendations.
Our study provides foundational evidence to:
• guide risk counseling,
• inform prevention strategies,
• refine expectations around age of onset, and
• support families navigating complex decisions in the absence of robust data.
Future prospective studies are necessary to more accurately estimate lifetime risk and define optimal timing for risk-reducing surgery.
References
1. Tischkowitz M, Huang S, Banerjee S et al. Small-Cell Carcinoma of the Ovary, Hypercalcemic Type-Genetics, New Treatment Targets, and Current Management Guidelines. Clin Cancer Res 2020; 26 (15): 3908–3917.
2. Young RH, Goodman A, Penson RT et al. Case records of the Massachusetts General Hospital. Case 8-2010. A 22-year-old woman with hypercalcemia and a pelvic mass. N Engl J Med 2010; 362 (11): 1031–1040.
3. Buys SS, Partridge E, Black A et al. Effect of screening on ovarian cancer mortality: the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Randomized Controlled Trial. JAMA 2011; 305 (22): 2295–2303.
4. Wens F, Hulsker CCC, Fiocco M et al. Small Cell Carcinoma of the Ovary, Hypercalcemic Type (SCCOHT): Patient Characteristics, Treatment, and Outcome-A Systematic Review. Cancers (Basel) 2023; 15 (15).
5. Jelinic P, Mueller JJ, Olvera N et al. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat Genet 2014; 46 (5): 424–426.
6. Kupryjanczyk J, Dansonka-Mieszkowska A, Moes-Sosnowska J et al. Ovarian small cell carcinoma of hypercalcemic type - evidence of germline origin and SMARCA4 gene inactivation. a pilot study. Pol J Pathol 2013; 64 (4): 238–246.
7. Le Loarer F, Watson S, Pierron G et al. SMARCA4 inactivation defines a group of undifferentiated thoracic malignancies transcriptionally related to BAF-deficient sarcomas. Nat Genet 2015; 47 (10): 1200–1205.
8. Longy M, Toulouse C, Mage P et al. Familial cluster of ovarian small cell carcinoma: a new mendelian entity? J Med Genet 1996; 33 (4): 333–335.
9. Witkowski L, Goudie C, Ramos P et al. The influence of clinical and genetic factors on patient outcome in small cell carcinoma of the ovary, hypercalcemic type. Gynecol Oncol 2016; 141 (3): 454–460.
10. Hansford JR, Das A, McGee RB et al. Update on Cancer Predisposition Syndromes and Surveillance Guidelines for Childhood Brain Tumors. Clin Cancer Res 2024; 30 (11): 2342–2350.



