
Welcome to the Gene Scene! Each week, we will explore a gene from the ACMG Secondary Findings list—genes identified by the American College of Medical Genetics and Genomics as having clear, actionable health implications. These genes are included because they’re linked to serious but preventable or manageable conditions when identified early. Here, we focus on the condition that led to the gene’s inclusion on the list, providing clear, relevant information that supports your clinic. To subscribe to the Gene Scene, contact your local GSL or send a request to info@ambrygen.com.
To access the Gene Scene archives, visit our blog.
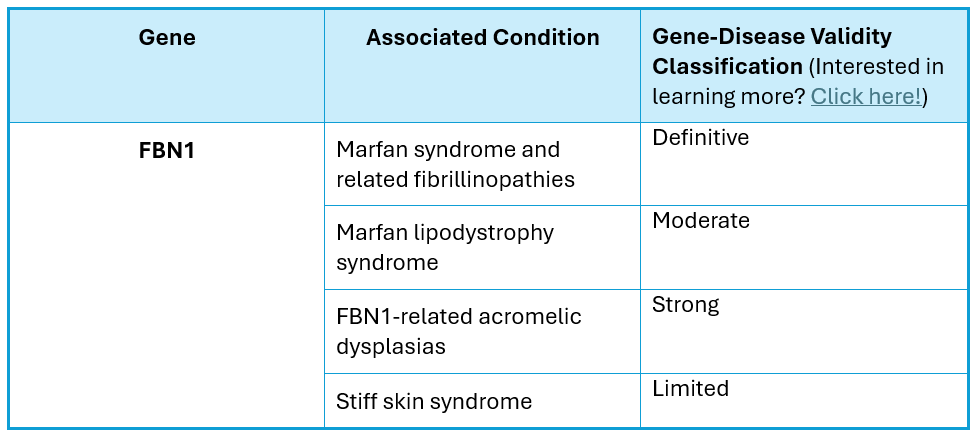
Clinical Phenotype Summary:
The FBN1 gene (NM_000138.4), which contains 65 coding exons and is located on chromosome 15q21, encodes the fibrillin-1 protein. Pathogenic variants in this gene are known to cause Marfan syndrome and related fibrillinopathies, Marfan lipodystrophy syndrome, and a spectrum of FBN1-related acromelic dysplasias, all of which are autosomal dominant conditions that may occur de novo. Classic Marfan syndrome is a systemic disorder of the connective tissue with features including skeletal, ocular, and cardiovascular involvement and is the most common of the fibrillinopathies (Loeys, 2010).
FBN1-Associated Marfan Syndrome (OMIM_154700) is characterized by:
• Aortic root dilatation Z-score ≥ 2
• Ectopia lentis
• Ghent II systemic findings, which may include a combination of:
o Cardiovascular features include mitral valve prolapse and acute dissection of the ascending aorta, which is usually the result of a slowly progressive aortic dilatation.
o Skeletal features, such as characteristic body habitus of tall stature, long and narrow limbs, a long and narrow head shape, scoliosis and pectus excavatum/carinatum. Individuals also have increased joint mobility.
o Skin findings
o Ocular findings (myopia, retinal detachment, and/or an acute glaucoma evoked by lens luxation)
o Facial features
• Other clinical features of Marfan syndrome include spontaneous pneumothorax, dural ectasia, and crowded teeth.
Clinical manifestation of Marfan syndrome is variable ranging from isolated features to a severe and rapidly progressive early onset presentation involving multiple organ systems which may be fatal early in life. However, the majority of individuals survive into mid- to late adulthood, and with proper management, the life expectancy of affected individuals approximates that of the general population (Faivre, 2007; Loeys, 2001; Loeys, 2004; Loeys, 2010). Clinical diagnosis of Marfan syndrome can be challenging due to the high phenotypic variability, however, it is often based on the presence of minor and major clinical criteria established as by the Ghent nosology (Loeys, 2010).
Both loss of function and variants reported to cause dominant negative effects have been reported as the mechanisms of disease for Marfan syndrome and related fibrillinopathies (Comeglio P et al., 2002; Regalado ES et al., 2016; Milewicz DM et al., 2021).
Marfan lipodystrophy syndrome (OMIM_616914) is characterized by:
• Congenital lipodystrophy
• Progeroid appearance
• Hyperextensible joints
• Arachnodactyly
• Myopia
• Other features seen in a minority of patients include lens dislocation, mitral valve prolapse, and/or aortic root dilatation.
De novo variants occurring in or affecting the penultimate exon have been reported as disease-causing for Marfan lipodystrophy syndrome (Takenouchi T et al., 2013; Passarge E et al., 2016).
FBN1-related acromelic dyplasias (including Weill-Marchesani syndrome (OMIM_608328), geleophysic dysplasia (OMIM_614185) and acromicric dysplasia (OMIM_102370) are characterized by:
• Severe short stature
• Brachydactyly
• Stiff joints
• Pseudomuscular build
• Thickened skin
• Distinguishing features of WMS are ocular anomalies, including ectopia lentis
• Distinguishing features of geleophysic dysplasia include progressive cardiac valvular thickening, distinctive facial features, radiologic findings, toe walking, respiratory insufficiency, and lysosomal-like storage vacuoles in various tissues.
• Acromicric dysplasia is less severe than geleophysic dysplasia with distinguishing features including hoarse voice and mild facial dysmorphism
Mechanism of disease for FBN1-related acromelic dysplasias is unknown (Le Goff C et al., 2011; Sakai LY et al., 2019; Stanley S et al., 2021).
Unique Considerations:
The presentation of Marfan syndrome can have clinical overlap with other Thoracic Aortic Aneurysm related disorders. GeneReviews has a helpful chart comparing the clinical features of Marfan Syndrome, Loeys-Dietz Syndrome, and Shprintzen-Goldberg Syndrome.
Clinical Resources:
Ambry’s Positive Result resource for TAAD related disorders
Ambry Knows Genes:
Scientific Posters:
ACMG 2023: Meghan Towne, MS, CGC: Variant reclassification in a large cardiogenetic testing cohort: The importance of disease-specific variant classification criteria
Citations:
• Comeglio P, et al (2002) The British journal of ophthalmology 86(12):1359-62 PMID: 12446365
• Dietz, H. (2001) GeneReviews. PMID: 20301510
• Faivre L., et al (2007) American Journal of human genetics. 81(3):454-66 PMID: 17701892
• Loeys, BL, et al (2010) Journal of medical genetics 47(7):476-85 PMID: 20591885
• Loeys, B, et al (2004) Human Mutation 24(2):140-6 PMID: 15241795
• Loeys B, et al (2001) Archives of Internal medicine 161(20):2447-54 PMID: 11700157
• Milewicz DM et al (2021) Nature reviews. Disease primers 7(1):64 PMID: 34475413
• Passarge E, et al (2016) European journal of human genetics 24(9):1244-7 PMID: 26860060
• Regalado ES, et al (2016) Clinical genetics 89(6):719-23 PMID: 26621581
• Sakai L, et al (2019) Matrix biology : journal of the International Society for Matrix Biology PMID: 30219651
• Stanley S (2021) Annals of the New York Academy of Sciences 1490(1):57-76 PMID: 32880985
• Takenouchi (2013) American journal of medical genetics. 161(12):3057-62 PMID: 24039054
• Le Goff C, et al (2011) American journal of human genetics 89(1):7-14 PMID: 21683322
Ambry Genetics Gene-Disease Validity Scheme
Each week, we explore a gene from the ACMG Secondary Findings list—genes identified by the American College of Medical Genetics and Genomics as having clear, actionable health implications. These genes are included because they’re linked to serious but preventable or manageable conditions when identified early.
To learn more about the ACMG Secondary Findings list, click here.
To read all previous Gene Scene emails, click here.



