
Welcome to the Gene Scene! Each week, we will explore a gene from the ACMG Secondary Findings list—genes identified by the American College of Medical Genetics and Genomics as having clear, actionable health implications. These genes are included because they’re linked to serious but preventable or manageable conditions when identified early. Here, we focus on the condition that led to the gene’s inclusion on the list, providing clear, relevant information that supports your clinic. To subscribe to the Gene Scene, contact your local GSL or send a request to info@ambrygen.com.
To access the Gene Scene archives, visit our blog.
GENE SCENE SPOTLIGHT: This gene is not on the ACMG Secondary Findings List. However, given the established gene-disease associations across multiple conditions and different mechanisms of disease, we wanted to spotlight this gene.
Clinical Phenotype Summary:
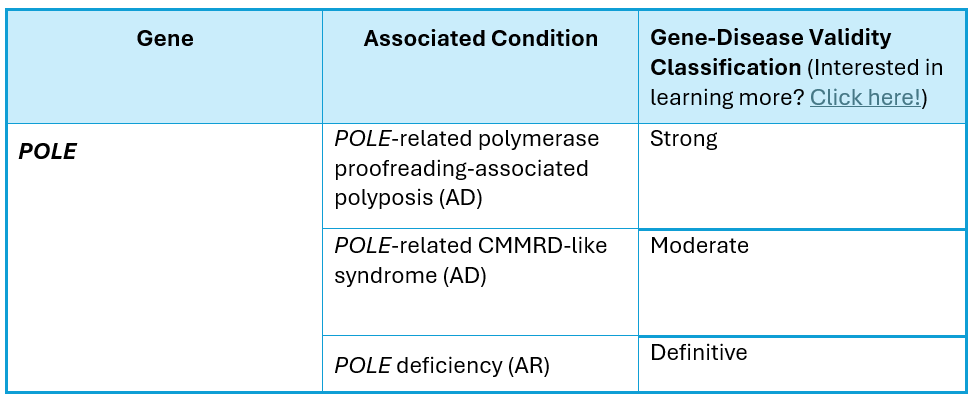
The POLE gene (NM_006231.2) is located on chromosome 12q24.33, encodes the DNA polymerase epsilon catalytic subunit A protein, and contains 49 coding exons. Pathogenic variants in this gene have been associated with POLE-related polymerase proofreading-associated polyposis (PPAP), which is inherited in an autosomal dominant fashion, POLE-related constitutional mismatch repair deficiency-like (CMMRD-like) syndrome, which is an autosomal dominant condition that generally occurs de novo, and POLE deficiency, which is inherited in an autosomal recessive fashion.
POLE-related PPAP is characterized by:
● Significantly increased risk (cumulative lifetime risk of 20% or greater) for polyposis and/or colorectal cancer (Palles C, et al. (2022) Fam Cancer 21(2):197-209; Valle L, et al. (2014) Hum. Mol. Genet. 23(13):3506-12; Buchanan DD, et al. (2018) Genet Med 20(8):890-895).
● Pathogenic POLE variants have also been detected in individuals with endometrial cancer; however, current evidence is insufficient to support a clear increase in risk of additional tumors over that of the general population.
● The penetrance of POLE-related PPAP is incomplete, and variable expressivity is observed; therefore, cancer risks and phenotype will differ based on individual and family history.
POLE-related CMMRD-like syndrome is characterized by:
● Significantly increased risk for early onset (childhood or young adult) polyposis and/or colorectal cancer, cutaneous features such as cafe-au-lait macules, and brain tumors (Sehested A, et al. (2022) Hum Mutat 43(1):85-96; Mur P, et al. (2023) Genome Med 15(1):85).
● Mechanism of disease is unclear for POLE-related PPAP and CMMRD-like syndromes, with reported pathogenic variants being missense or in-frame variants restricted to the exonuclease (proofreading) domain of POLE (Mur, 2023).
POLE deficiency is characterized by:
● Intrauterine growth restriction, postnatal short stature, microcephaly, facial dysmorphism (including micrognathia, long thin nose, crowded dentition, wide neck, and/or small, low set posteriorly rotated ears), immunodeficiency, developmental delay, skeletal anomalies (micromelia, delayed bone age, and/or metaphyseal and epiphyseal dysplasia), adrenal insufficiency, and genitourinary anomalies in males (Logan CV, et al. (2018) Am J Hum Genet 103(6):1038-1044;Roberts ME, et al. (2022) Am J Med Genet A 188(10):3121-312).
● Biallelic loss of function has been reported as the mechanism of disease for POLE deficiency.
● Individuals of reproductive age are at 25% risk of having a child with POLE deficiency with each pregnancy when both biological parents have a loss-of-function pathogenic variant in POLE.
Unique Considerations:
Pathogenic variants in POLE are associated with autosomal recessive and autosomal dominant disease. In addition, the mechanism of disease differs between the conditions.
POLE deficiency:
● The mechanism of disease is loss-of-function.
POLE-related PPAP and CMMRD-like syndrome:
● The mechanism of disease is likely gain-of-function.
● Pathogenic variants are restricted to missense or in-frame variants in the exonuclease (proofreading) domain of POLE.
Clinical Resources:
Clinical Management Resource for POLE and Understanding Your Positive POLE Genetic Test Result
Clinical Management Resource for POLE and Understanding Your Positive POLE Carrier Genetic Test Result
Citations:
● Sehested A et al. Human mutation. 2022 Jan;43(1): 85-96. PMID 34816535
● Mur P et al. Genome medicine. 2023 Oct;15(1):85. PMID 37848928
● Roberts ME et al. American journal of medical genetics. Part A. 2022 Oct;188(10):3121-3125. PMID 35860951
● Mur P et al. Genetics in medicine. 2020 Dec;22(12):2089-2100. PMID 32792570
● Briggs S et al. The Journal of pathology. 2013 Jun;230(2):148-153. PMID 23447401
● Thiffault I et al. BMC medical genetics. 2015 May;16(1):31. PMID 25948378
● Logan CV et al. American journal of human genetics. 2018 Dec;103(6):1038-1044. PMID 30503519
● Palles C et al. Familial cancer. 2022 Apr;21(2):197-209. PMID 33948826
● Buchanan DD et al. Genetics in medicine. 2018 Aug;20(8):890-895. PMID 29120461
● Valle L et al. Human molecular genetics. 2014 Jul;23(13):3506-3512. PMID 24501277
● Palles C et al. Nature genetics. 2013 Feb;45(2):136-144. PMID 23263490
● Church JM. Diseases of the colon and rectum. 2014 Mar;57(3):396-397. PMID 24509466
Ambry Genetics Gene-Disease Validity Scheme
Each week, we explore a gene from the ACMG Secondary Findings list—genes identified by the American College of Medical Genetics and Genomics as having clear, actionable health implications. These genes are included because they’re linked to serious but preventable or manageable conditions when identified early.
To learn more about the ACMG Secondary Findings list, click here.
To read all previous Gene Scene emails, click here.



